汇聚连通各界科技人员,海纳百川,将知识和技能服务于有关行业,促科技与经济进步,直答理工网就是这样一个科研人员的社区有偿问答平台。
前言
Lammps是一款免费开源的分子动力学软件(官网:http://lammps.sandia.gov/)。windows和linux版本都有。网上的安装教程要么年代久远,要么省去了步骤,要么有多余的步骤。于是就有了这么一篇教程。下面所有操作我都用目前最新的安装包测试过了。用的是阿里云CentOS 5.8 64位的服务器。所有命令用红色字体表示,并加以说明。希望能给刚接触linux和lammps的同学一些帮助。
windows版本
我测试用的电脑系统为win7 32位。windows版本也是可以并行计算的,这里只演示串行版本。下载对应系统版本,然后默认安装。C:\Program Files\LAMMPS 32-bit 11Aug2017\bin目录下的
lmp_serial.exe是运行文件。

然后例子就开始运行了。
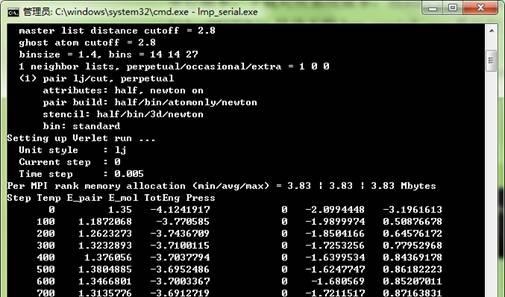
不熟悉linux的同学可以用windows版本来做一些测试。windows版本的lammps的安装和运行一般不会出现问题。
linux版本(串行)
这里测试我用的是阿里云CentOS 5.8 64位服务器,自己的集群跑满了不太方便测试。直接用的是root账户。串行和并行用同样的安装包就可以了。下面所有的安装包都放在/opt目录下。你可以根据自己的情况修改,只需要注意将后面的/opt路径改成你的安装路径。
??检查c、c 、fortran编译器
which gcc #c
which g #c
which gfortran #fortran
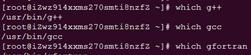
一般的linux系统都有这些编译器。
?? 上传,解压,编译
我下载的是lammps-stable.tar.gz,上传目录/opt。
cd /opt #切换目录
tar -zxvf lammps-stable.tar.gz #解压,具体文件名可能有变
cd /opt/lammps-11Aug17/src/STUBS #进入/STUBS目录,具体文件名可能有变
make #make
cd .. #切回/src目录
make serial #串行编译
?? 检查测试
查看/Src目录下是否生产了lmp_serial文件,这个就是执行文件,与windows下的类似。
同样的运行例子。
cd /opt/lammps-11Aug17/examples/KAPPA #进入例子目录
/opt/lammps-11Aug17/src/lmp_serial < in.heat #运行例子in.heat
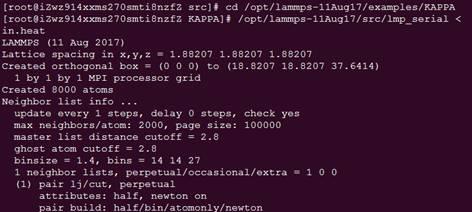
linux版本(并行)
准备fftw
、mpich、lammps安装包。检查g 、gcc、gfortran编译器。值得一提的是,fftw、mpich这两个软件在安装其他软件的时候也会用到,这里装好了以后可以直接用。
??fftw的安装
cd /opt
tar -zxvf fftw-3.3.6-pl2.tar.gz #解压
cd fftw-3.3.6-pl2 #进入目录
./configure –prefix=/opt/fftw3 –enable-float
#设置安装路径,这里我设置的是/opt/fftw3
make
make install
安装完成就可以把fftw-3.3.6-pl2文件夹和安装包删除了,安装好的fftw在/opt/fftw3目录下。
??fmpich的安装
cd /opt
tar -zxvf mpich-3.2.tar.gz
cd mpich-3.2
./configure –prefix=/opt/mpich3
make
make install
和fftw类似,mpich我设置安装在/opt/mpich3目录下。
?? 设置环境变量
.bashrc文件在你登陆账户的主目录下
cd #进入主目录
vi .bashrc #修改.bashrc文件
在文件后面加上这样两行
exportPATH=/opt/mpich3/bin:/opt/fftw3/bin:$PATH
exportLD_LIBRARY_PATH=/opt/mpich3/lib:/opt/fftw3/lib: $LD_LIBRARY_PATH
键盘输入i进入修改模式,然后输入上面的两行,注意,上面的路径要改成你自己安装的路径。或者直接下载.bashrc到本地,在windows下改好了再上传。
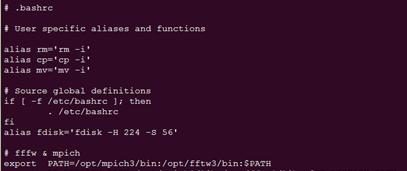
这样就添加好了,按键盘Esc键,然后输入:wq(保持并退出)。最后用命令source .bashrc环境变量生效。可以输入which mpirun 检查一下;
??Lammps并行编译
解压
cd /opt
tar -zxvf lammps-stable.tar.gz
最最关键的地方,修改/opt/lammps-11Aug17/src/MAKE目录下的Makefile.mpi文件。
cd /opt/lammps-11Aug17/src/MAKE
vi Makefile.mpi
??第一处编译器修改,将原来的mpicxx注释或删除
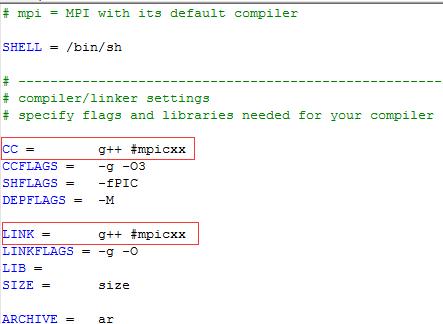
??第二处 mpich函数库添加,路径都要改成你安装mpich的路径
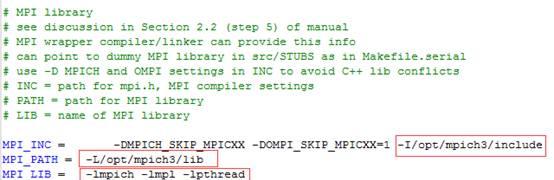
MPI_INC = -DMPICH_SKIP_MPICXX -DOMPI_SKIP_MPICXX=1 -I/opt/mpich3/include
MPI_PATH = -L/opt/mpich3/lib
MPI_LIB = -lmpich -lmpl -lpthread
??第三处fftw
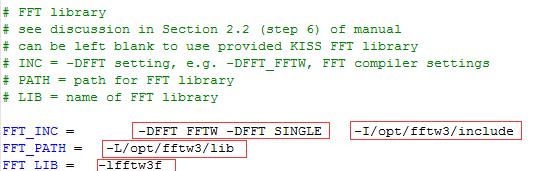 FFT_INC = -DFFT_FFTW -DFFT_SINGLE -I/opt/fftw3/include
FFT_INC = -DFFT_FFTW -DFFT_SINGLE -I/opt/fftw3/include
FFT_PATH = -L/opt/fftw3/lib
FFT_LIB = -lfftw3f
保存退出然后编译
cd /opt/lammps-11Aug17/src
make mpi
等编译完成,src目录下就会生产lmp_mpi文件。
?? 测试
这里我们可以设置lmp_mpi的环境变量,设置环境变量的好处是,每次计算就不用输入lmp_mpi的完整路径了。
cd /opt/lammps-11Aug17
cp src/lmp_mpi . #把lmp_mpi复制到lammps主目录下
cd #回到用户主目录
vi .bashrc
在.bashrc加上lmp_mpi的路径,和前面的fftw类似。
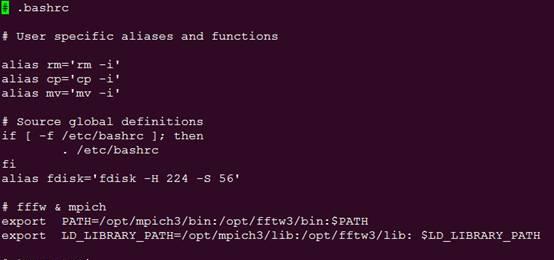
source .bashrc
cd /opt/lammps-11Aug17/examples/KAPPA
mpirun -np 8 lmp_mpi < in.heat
上面的8代表用8个核计算。
额外打开一个终端,输入top就可以看到lmp_mpi进程。如果不设置lmp_mpi的环境变量,上面的运行命令就应该是mpirun -np 8 /opt/ lammps-11Aug17/lmp_mpi < in.heat。
直答理工网招聘校园兼职学生!